

The high-stakes world of biopharmaceuticals, the distance between a state-of-the-art facility and a multi-million dollar Warning Letter is thinner than a sterile filter.
As we navigate the regulatory landscape of 2026, the mantra has shifted from passing the inspection to a sustained state of control.
Whether you are breaking ground on a greenfield site or pivoting to a modular cell-and-gene therapy suite, achieving regulatory readiness is no longer a final hurdle; it is the track you run on.
Gone are the days when quality was inspected into a product at the end of the line. In 2026, regulatory bodies like the FDA and EMA demand that compliance be woven into the very architecture of your plant.

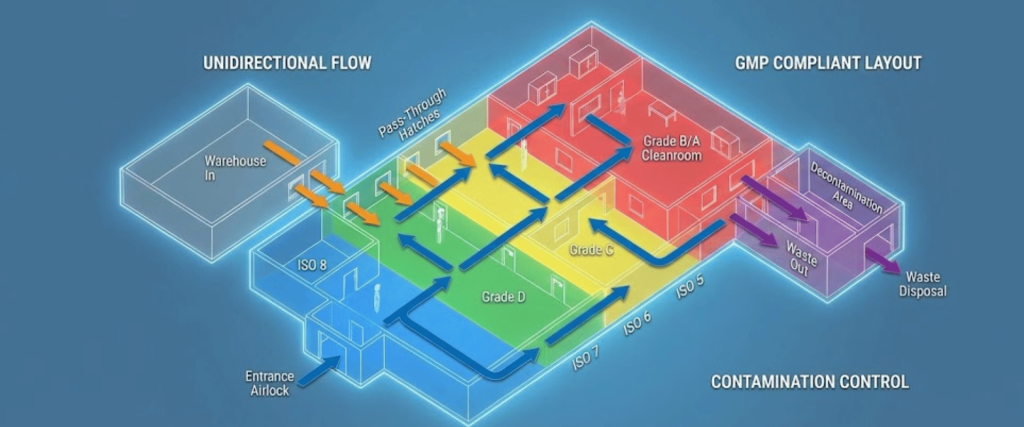
Your facility layout is your first line of defense against cross-contamination.
In 2026, an audit is essentially a forensic deep-dive into your data. To be ready, your plant must adhere to ALCOA++ principles (Attributable, Legible, Contemporaneous, Original, Accurate, plus Complete, Consistent, Enduring, and Available).
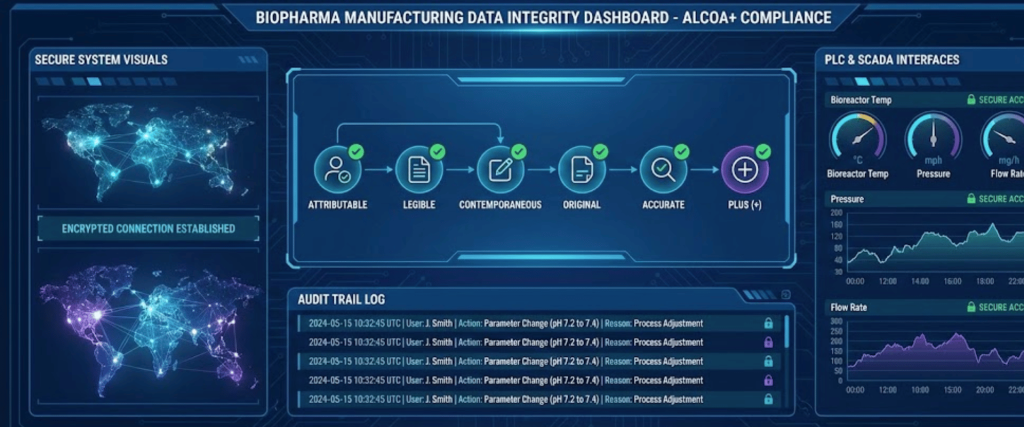
Pro Tip: If your data isn’t timestamped in real-time by a validated system, in the eyes of an inspector, it didn’t happen.
Achieving readiness is a marathon, not a sprint. Breaking it down into phases ensures that no critical validation step is missed.
Before the first steel beam is placed, you must define your Regulatory Target Profile.

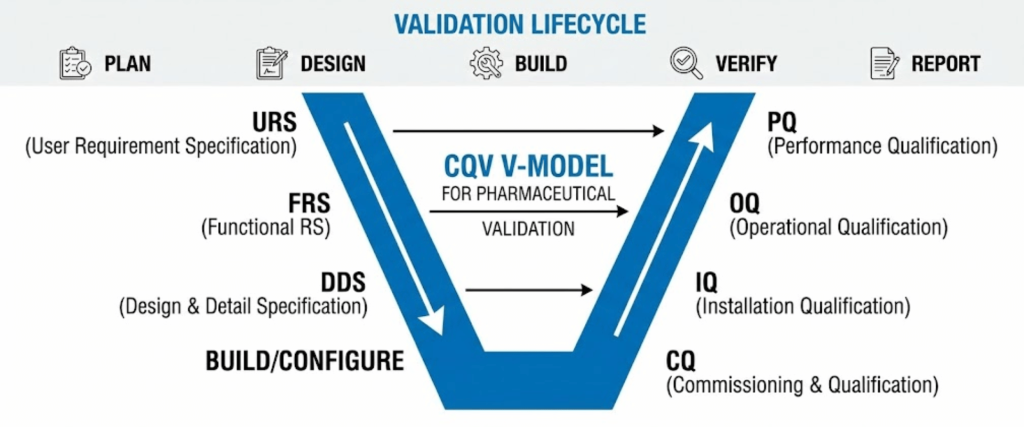
The V-Model remains the gold standard for ensuring that what you designed is actually what you built.
Never let the FDA be your first auditor. Conduct a rigorous internal mock inspection six months before your intended launch. Focus on the story your documentation tells.

Can you trace a single vial back to the specific batch of raw media used, the sensor’s calibration record, and the operator’s training certificate?
The Factory of the Future is here, and it brings new regulatory questions.

Use this checklist to gauge your plant’s maturity.
| Category | Requirement | Status |
|---|---|---|
| Personnel | Are 100% of staff trained on the latest cGMP and specific SOPs? | [ ] |
| Validation | Is the Master Validation Plan (MVP) signed off and up to date? | [ ] |
| Suppliers | Have all critical Tier 1 suppliers been physically or virtually audited? | [ ] |
| Data | Are all PLC/SCADA systems compliant with 21 CFR Part 11? | [ ] |
| Safety | Are containment systems (Isolators/RABS) validated for the specific HPAPI? | [ ] |
The most successful bio-pharma plants in 2026 treat regulatory readiness as a byproduct of operational excellence.
When your processes are robust, your data is transparent, and your culture is quality-first, the inspection becomes a mere formality, a chance to show off your hard work rather than a week of anxiety.
Would you like me to draft a more detailed Mock Audit checklist specifically tailored to the latest 2026 FDA/EMA harmonized standards?
Compliance-by-Design integrates regulatory requirements directly into the engineering and architectural phase of a facility. By addressing issues like unidirectional flow, airlock transitions, and closed-system processing during the design stage, Regulatory Readiness companies avoid the validation traps that lead to costly retrofitting later. In 2026, CbD is the primary strategy for ensuring that a plant is not just operational, but audit-ready from the moment the first piece of equipment is installed.
With the industry moving toward Paperless Manufacturing (Industry 4.0), data integrity is the top focus for inspectors. Facilities must adhere to ALCOA++ principles, ensuring all data is Attributable, Legible, Contemporaneous, Original, and Accurate.
While a major mock audit is critical six months before a formal inspection (such as a Pre-Approval Inspection or PAI), the 2026 standard is Continuous Readiness. Leading firms now conduct Internal Pulse Checks every quarter. These smaller, Regulatory Readiness-focused audits target specific high-risk areas like cleaning validation or personnel training records to ensure that the facility maintains a constant state of control, rather than scrambling to prepare only when an inspection is announced.
