

In automated pharmaceutical packaging, the air is as much a raw material as the active ingredients themselves.
Since many life-saving drugs are sensitive to microbial contamination or particulate matter, the environment where they are sealed must be strictly controlled.
Validation is the documented evidence that a system in this case, the air filtration and distribution within a packaging machine consistently performs according to specified standards.
Without robust validation, even the most advanced automated systems risk introducing contaminants that can lead to batch recalls, regulatory fines, and, most importantly, compromised patient health.
At the heart of sterile air quality are High-Efficiency Particulate Air (HEPA) filters. In an automated setup, these filters must provide a Grade A environment (ISO 5) at the point of fill.
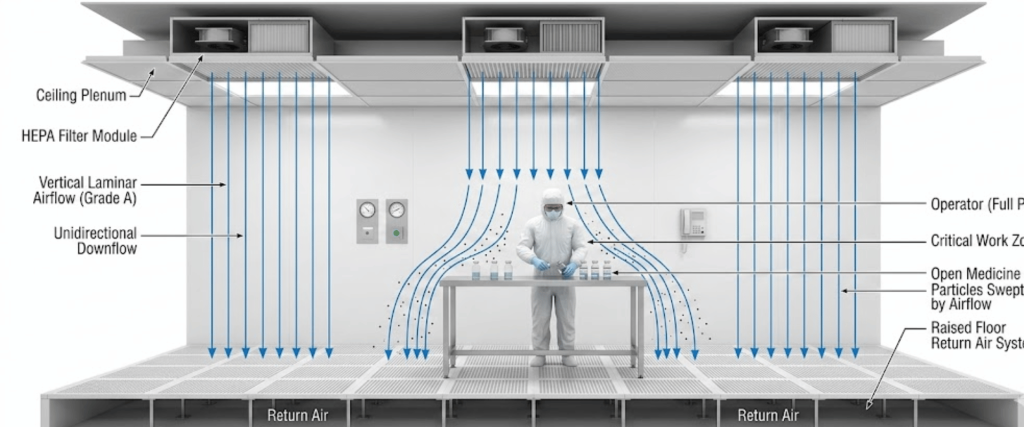
Pharmaceutical packaging must adhere to international guidelines to ensure global compliance. The two primary frameworks.

The following table outlines the requirements for different zones within a pharmaceutical facility.
| Grade | ISO Equivalent | Particle Limit (≥0.5 μm/m³) | Recommended Use |
|---|---|---|---|
| Grade A | ISO 5 | 3,520 | High-risk operations (filling, sealing) |
| Grade B | ISO 5 (at rest) | 3,520 | Background environment for Grade A |
| Grade C | ISO 7 | 352,000 | Preparation of solutions for filtration |
| Grade D | ISO 8 | 3,520,000 | Handling of components after washing |
Validating sterile air is not a one-time event; it is a continuous cycle of testing and documentation.
This stage ensures that the air handling units (AHUs), ductwork, and filters are installed correctly according to the manufacturer’s specifications. It involves checking model numbers, duct integrity, and sensor placements.

During OQ, the system is tested to see if it performs as intended under worst-case scenarios.

PQ is conducted during actual production runs. This confirms that the air remains sterile even when the automated machinery is moving, generates heat, and creates potential turbulence.

Automation brings efficiency, but it also introduces unique variables.
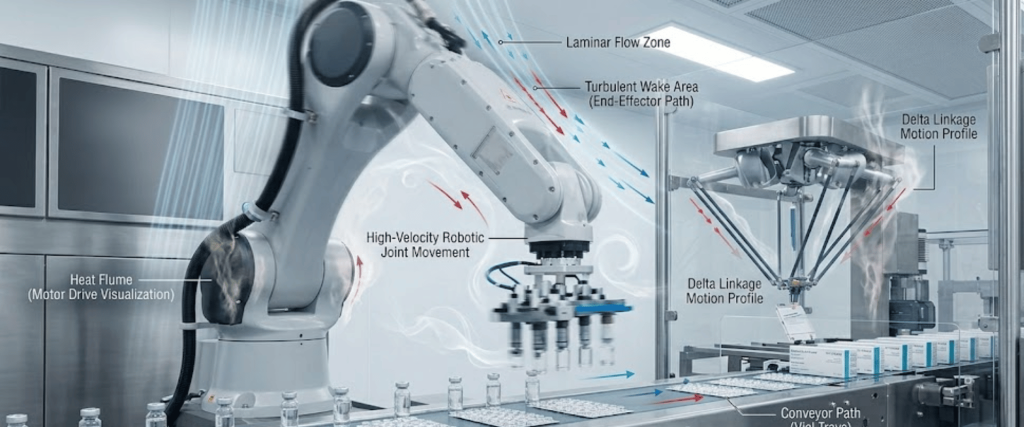
Modern pharmaceutical lines are moving away from manual sampling toward Continuous Environmental Monitoring Systems (CEMS).

Validating sterile air quality in automated pharmaceutical packaging is a blend of rigorous engineering and strict regulatory adherence.
By focusing on HEPA integrity, maintaining pressure differentials, and conducting thorough PQ cycles, manufacturers can guarantee the safety and efficacy of their products.
As automation continues to evolve, the tools we use to validate the invisible environment of air will remain the most critical component of the production line.
Sterile air systems in pharmaceutical packaging should undergo full re-qualification at least every 6 to 12 months. However, critical parameters like pressure differentials and particle counts should be monitored continuously during every production shift to ensure ongoing compliance with ISO 5 standards.
A Smoke Study, or airflow visualization, is used to prove that the laminar airflow remains Uninterrupted even when automated robotic arms or conveyors are moving. It ensures that no dead zones or turbulence exist that could potentially trap contaminants near the open drug product.
If air quality exceeds the allowed particle limits, the production line must be stopped immediately. This triggers a Root Cause Analysis (RCA) and usually requires the quarantine of all products packaged since the last successful test. Regular validation prevents these costly batch rejections and regulatory warnings.

